Home
- Overview
- Installation
- Geometry optimization
- Structure Generation
- Setting Up Your Input Files
- Setting up the submit script and submitting jobs to the queue
- Viewing your output file
- Running a growing string calculation
- General overview of the DE-GSM and SE-GSM
- Getting started
- Double-ended Growing String Method (DE-GSM)
- Single-ended Growing String Method (SE-GSM)
- Managing QSub Files for DE-GSM/SE-GSM
- Analyzing Output
- Advanced Options
- Troubleshooting and FAQS
- References
The Growing String Method (GSM) is a reaction path (RP) and transition state (TS) finding method that develops a RP by iteratively adding new nodes and optimizing them until a complete RP with a TS and a stable intermediate on each side of the string are present. The string is represented by a discretized set of structures along the RP connecting the reactant and product geometries and its formation can be initiated from a single initial geometry or a pair of initial and final structures. By incremental addition of new nodes, GSM rapidly leads to a reasonably well converged RP since it avoids placing nodes at high-energy regions of the potential energy surface. GSM has been shown to outperform competing chain-of-states methods that generate and place all the nodes at once because it avoids optimization of high-energy structures in the middle of the RP.
This method operates through three overall phases: growth, optimization, and exact TS search. The growth phase builds the string representing the RP by sequential node addition and optimization until a reasonably converged path is formed. When the string formation is complete, the optimization phase refines the RP, which also creates a guess-geometry for the exact TS search phase. At the last phase, a saddle point search follows the Hessian eigenvector of the TS node that most closely resembles the reaction path, resulting in a reliable TS optimization. In this phase, the non-TS nodes of the RP continue to be optimized, resulting in a simultaneous formation of the exact TS and minimum energy RP.
create an anchor To compile:
- Set FC and LINKERFLAGS in Makefile --I have used Intel 12 and 13 compilers
- type: make
- copy gfstringq.exe to run directory
- copy nwchem.py to your ase/calculators/ directory
To run gfstringq.exe:
- Setup grad.py by setting scratch directory to local scratch --default is $PBSTMPDIR
- Set XC and basis in grad.py --default B3LYP/6-31G**
- Check inpfileq for string method settings
- To run: a. NWChem must be available at command line (e.g. source setnw) b. export OMP_NUM_THREADS=1 c. to execute: ./gfstringq.exe jobnumber numberofcores > scratch/paragsmXXXX d. initialXXXX.xyz and ISOMERSXXXX must be present in scratch
- example/qmakeg can create a queuing script in scratch/ called go_gsm_dft.qsh --add "#PBS -t jobnumber1,jobnumber2,..." at the top of go_gsm_dft.qsh
Analysis:
- stringfile.xyzXXXX contains the reaction path and TS --variants on this file w/"g" at the end are growth phase strings --"fr" at the end is a partial Hessian analysis, showing the first 3 vibrational modes --comment lines are energies relative to first structure in kcal/mol
- paragsmXXXX contains the optimization output
- ./status shows the current state of the various runs
Examples in example/ directory:
0007: This is a Diels-Alder reaction. initial0007.xyz contains input for SE-GSM (SSM) or DE-GSM (GSM) 0076: H2 addition to SiH2. initial0076.xyz contains SSM and GSM input.
#Geometry optimization The geometry optimization for molecules in gas phase is divided into 4 steps. The procedure is explained using the acetic acid example.
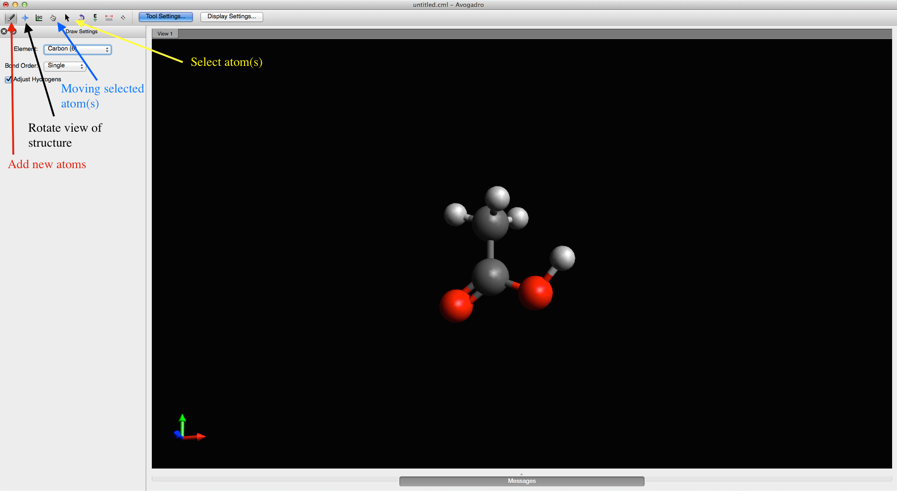
###Step 1: Structure Generation In order to run reaction path finding calculations, you first need to generate the input structure(s). To do this, you need a visualization software such as Avogadro. When using Avogadro (or any other type of visualization software):
- Draw your structure using the drawing tools
- Check that all atoms are the correct element
- Check that all atoms are connected to the appropriate number of hydrogen atoms (or other atoms)
- Check the bond order for all connections
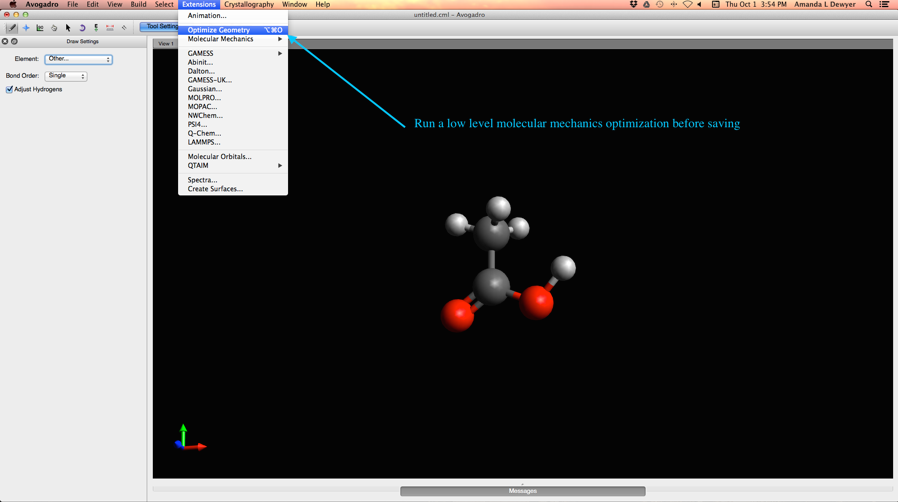
- Run a quick low level geometry optimization (see below)
- Double check structure to make sure it looks like a good guess at the starting structure for your reaction of interest (e.g. sometimes hydrogen atoms won't be oriented correctly, or the conformation of a ring structure may change in an undesirable way)
- Save your file in .xyz format
See images below for more details:
Avogadro Tools
Optimize Structures Before Saving
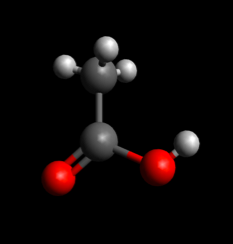
Double check structures before saving
Poor starting structure guess
Good starting structure guess
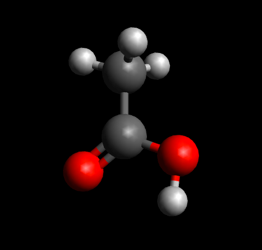
Both structures above are optimized; the bottom one is lower in energy though.
Save Structure in .xyz format
The .xyz file you save holds the Cartesian coordinates that define the structure you generated. These coordinates will be used for DFT geometry optimizations using QChem.
###Step 2: Setting Up Your Input Files In order to run an optimization you need the following files (sample files can be found here):
- Cartesian coordinates of the input structure (generated above)
- q001.template.inp
- submitall.qsh
- getOptEnergy
- makeXYZ
- Knowledge of the appropriate levels of theory to run your calculation at:
- Density functional (ex. B3LYP)
- Exchange Functional
- Correlation Functional
- Basis set (ex. 6-31+G*)
- Restricted or Unrestricted DFT
- Effective Core Potential (ECP) for heavy atoms
- The charge of your structure
- The spin multiplicity of your structure Once you have the above files and details about your calculations you can generate your input file. The file "q001.template.inp" is a template file for optimization calculations.
- Density functional (ex. B3LYP)
- Exclamation point (!) indicates that the line is commented out (QChem will not read the line).
$rem
JOBTYPE #Tells QChem which type of job is being ran
EXCHANGE #Defines the density functional being used
! CORRELATION #Specifies the use of an optional correlation functional
! UNRESTRICTED #Defines if calculation uses restricted or unrestricted DFT
SCF_ALGORITHM #Specifies the SCF algorithm used in QChem
SCF_MAX_CYCLES #max number of SCF cycles before convergence failure
BASIS #Indicates the basis set being used
ECP #Effective Core Potential being used on heavy atoms
WAVEFUNCTION_ANALYSIS
GEOM_OPT_MAX_CYCLES #max number of geom opts before the optimization fails
scf_convergence #convergence threshold for SCF cycles
SYM_IGNORE #Indicates if symmetry is ignored
SYMMETRY #Indicates if symmetry is turned on
molden_format #Prints xyz coordinates in molden format in the output file
$end
$molecule
0 1 #Charge | Multiplicity
#Cartesian coordinates of structure goes here
$endTo generate your input file, copy this file to a new one with the designated name of your choice. Then edit your new file and fill in the appropriate levels of theory etc. for your calculation.
###Example: Acetic Acid geometry optimization run using the following levels of theory B3LYP density functional, restricted DFT, LANL2DZ basis set, neutral molecule, spin multiplicity of 1.
$rem
JOBTYPE OPT
EXCHANGE B3LYP
! CORRELATION PBE
! UNRESTRICTED TRUE
SCF_ALGORITHM diis
SCF_MAX_CYCLES 150
BASIS LANL2DZ
ECP LANL2DZ
WAVEFUNCTION_ANALYSIS FALSE
GEOM_OPT_MAX_CYCLES 300
scf_convergence 6
SYM_IGNORE TRUE
SYMMETRY FALSE
molden_format true
$end
$molecule
0 1
O -1.93776 -0.72249 0.00000
C -0.83158 -0.21089 -0.00000
O 0.28198 -0.96655 -0.00000
C -0.52791 1.25073 -0.00000
H -1.46497 1.81470 -0.00000
H 0.03663 1.51185 -0.89846
H 0.03663 1.51185 0.89846
H -0.05498 -1.88701 0.00000
$end###Step 3: Setting up the submit script and submitting jobs to the queue
Once your input file is generated, you can run optimizations using QChem by submitting jobs to the queue through using the “submitall.qsh” script as shown below:
#PBS -t 2
#PBS -l nodes=1:ppn=2 -l pmem=2000MB -l walltime=24:00:00
ID=`printf “%0*d\n” 3 ${PBS_ARRAYID}`
#i=$PBS_ARRAYID
source /export/zimmerman/paulzim/qchem/qchemjan42013c/paul.set.local
cd $PBS_O_WORKDIR
name=`ls q$ID*.inp`
qchem -np 2 $name $name.out-
#PBS –t: Indicates the jobs you want to run. It refers to the input file number (q002.methane.inp). You can specify a range or series of files. (ex. “#PBS –t 2-10” or “#PBS –t 2,5,7”) -
ID=`printf “%0*d\n” 3 ${PBS_ARRAYID}`: Indicates the ID of the file by looking at the 3 digits indicated at in the input file. -
source /export/zimmerman/paulzim/qchem/qchemjan42013c/paul.set.local: Sources QChem. Make sure to use the correct path based on the queue you are submitting your jobs to (zimmerman vs. guest, …) -
name=`ls q$ID*.inp`: Sets the file name -
qchem -np 2 $name $name.out: Indicates the number of processors (np) QChem is using. np in this line needs to match ppn in#PBS –lline. For more information on submitting jobs to the queue see the “Workstation Setup” (TODO) tutorial.
###Step 4: Viewing your output file
Once your optimization calculations finish, you then need to look at the output. Output files will have the .out extension. QChem will show a message at the end of the output file indicating successful optimization. You can then run the getOptEnergy and makeXYZ executables to obtain the energy of the structure in Hartrees and xyz file of the final optimized geometry, respectively. Once you have completed the geometry optimizations you can use it to search for reaction paths.
#Running a growing string calculation
###General overview of the DE-GSM and SE-GSM
As this is most likely the first time you have used the growing string methods, we would encourage you to spend a moment to learn how the software operates. Please see the most recent literature papers detailing the functionality of the GSM software ((Zimmerman JCTC... FILL THIS IN)). As these are fairly technical, please also discuss the program with labmates. We have included a very general overview of the method here.
The growing string method is a coordinate driving transition state search algorithm developed for use in the Zimmerman group. It is a state-of-the-art transition state (TS) finding method that should operate without much user interaction. Depending on the method the user input varies: for the single ended method the starting structure and bonding changes are required, for the double ended method the start and end point are required (no specific bonding changes need to be input by the user).
The method takes the input information and generates a string of “nodes”. These nodes represent various geometric conformations along the reaction tangent (the path that the reaction is taking) (Figure 1).
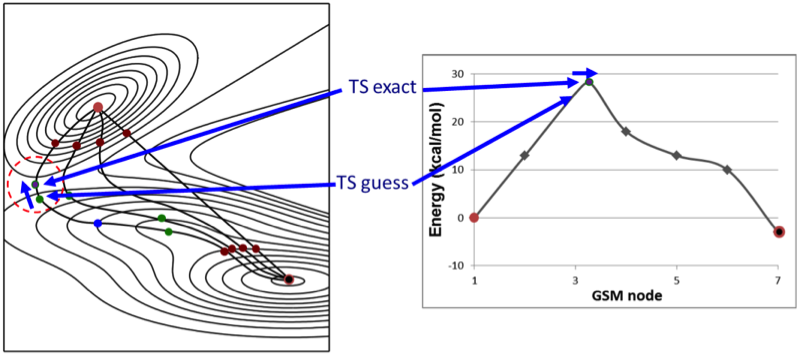
Figure 1. The left picture shows the addition of nodes on the string connecting the starting and end point of an example DE-GSM run. As more nodes are added and subsequently optimized, the string begins to converge toward the transition state. Note how the nodes in the energy diagram on the right increase in energy as they get closer to the transition state. This is considered a good profile for evaluation of a single elementary step. In the final portions of a growing string run, the software takes the highest energy node and optimizes the structure along the reaction path toward the transition state.
As the string is evaluated all of the nodes are optimized repeatedly until the convergence threshold (in inpfileq) is met. At this point the software will evaluate the “exact transition state”. If successful the run will complete and return the output to the user. No further optimization of the transition state is needed; however, some groups who use this software utilize more traditional methods to obtain a more exact transition state. Typical results with the redundant optimization have been only marginal improvements of ~1 kcal mol-1 (don’t cite this, it is an estimate).
The next sections will outline specific user interaction with the software, how to setup the input file, and common mistakes. Obviously if there are ever any questions a fellow lab member can be contacted for assistance.
###Getting started
####Should I use double-ended GSM (DE-GSM) or single-ended GSM (SE-GSM)?
In general, ask yourself the following questions before choosing a particular string method:
- Do you know the end point (final product) of the string you are searching for?
- Do you have the structure optimized for the product of your reaction?
- Are the coordinates of the reaction (the bonds/angles/torsions) difficult to define?
- Are you searching for a single product with a known final conformation?
If you answered yes to any of these questions, then the best bet is probably the double ended growing string method. For all other cases we would advise attempting the single ended method first. While the success rate (finding the final product and transition state) is a bit less reliable for the single ended method, the results tend to be obtained faster and with less input from the user.
Finally, I will end this introduction by saying that if you have any questions about any of the information found in this tutorial, please feel free to contact another senior member of the group for a more detailed explanation.
Coming together is a beginning; keeping together is progress; working together is success. –Henry Ford
###Double-ended Growing String Method (DE-GSM)
####Important notes moving forward:
In cases where one has elected to use the DE-GSM, you must be certain about the numbering of atoms in the XYZ file. To elaborate, your input parameters (the two starting XYZ files containing the starting material and the product of the transformation) need to have the atoms arranged in the same order. If this is not done correctly, the DE-GSM will not locate the transition state you are looking for.

Obtaining pairs of reactant/product geometries with the correct (matching) atomic arrangement can be achieved in Avogadro and other molecular editors. One can achieve this in Avogadro by adding/removing bonds from the starting structure that correspond to the product/intermediate structure. It is important to avoid adding/deleting atoms as this reorders the atoms within the XYZ file. Lastly, using the force field optimization option in Avogadro should give you an approximate geometry for the product (with the correct order of atoms in the XYZ file) that can be optimized in QChem.
Continue reading once you are certain that you have obtained the correct starting structures. There is no shortcut for rearranging XYZ files yet (maybe you can develop one [in C++ preferably]!), for now just do it by hand. This can be quite tricky but is vital for the DE-GSM to function. If you are deterred by this prospect, skip ahead to the SE-GSM section.
####Setting up the DE-GSM
First copy a template directory containing the most recent version of the GSM along with the necessary files into your home directory (or wherever you plan on running the DE-GSM). A number of files will be created along with the final stringfile, make sure that your directory is specific enough that the files generated will be easy to distinguish.
A template directory can be found in:
/export/zimmerman/ipendlet/GSM_Files/GSM_template
Please note that the DE-GSM in this directory might not be the most updated version. Check the directory update date by using the ls –la command. The only file that absolutely needs to be updated is the gfstringq.exe file, but you may also need a recent version of “inpfileq” to use new functionalities added to the growing string.
Copy the directory to where you would like to run the job and create your queue submission script through the following command:
$ ./qmakeg
This should generate a qsub file in the scratch directory named go_gsm_dft.qsh. We will come back to this file shortly. Also, one can change the qmakeg file to produce the disired qsub file in the scratch directory.
Take your optimized reactant and product files and put them together through the “cat” command:
$ cat test1.xyz test2.xyz >> scratch/initial0001.xyz
Or by using a text editor such as vim. Additional input files are named sequentially: initial0002.xyz, initial0003.xyz … etc. The test1.xyz file is a stand-in for your starting molecule file.
Because DE-GSM invokes QChem for the optimization of nodes along the string that represents the reaction path, it is important to indicate the correct charge and multiplicity for the structures provided in the “initial####.xyz” files as well as the level of theory (functional/basis set). The template directory provided contains a file called “qstart” where you can input the charge, multiplicity, and level of theory. This file uses the same format as QChem, so you should know where to input the charge, multiplicity, density functional, and basis set.
From here follow the instructions above outlining the use of the qsub file. All of the default settings should be fine as long as the directories are copied. For more advanced functionality and debugging please consult the advanced section below.
###Single-ended Growing String Method (SE-GSM)
First copy a template directory containing the most recent version of the growing string (gfstringq.exe) along with the necessary files into your home directory (or wherever you plan on running the SE-GSM). A number of files will be created along with the final stringfile, make sure that your directory is specific enough that the files generated will be easy to distinguish.
A template directory can be found in:
/export/zimmerman/ipendlet/GSM_Files/SSM_template
Please note that the SE-GSM executable (gfstringq.exe) in this directory might not be the most updated version. Check the directory update date by using the ls –la command. The only file that absolutely needs to be updated is the gfstringq.exe file.
Copy the directory to where you would like to run the job and create your queue submission script through the following command:
$ ./qmakeg
This should generate a qsub file in the scratch directory named go_gsm_dft.qsh. We will come back to this file shortly.
Take your input files and put them together through the “cat” command:
$ cat test1.xyz >> scratch/initial0001.xyz
Or by using a text editor such as vim. Additional input files are named sequentially: initial0002.xyz, initial0003.xyz … so on. Note that each "initial" file will only have one set of xyz coordinates since the SE-GSM only requires the reactant and search coordinates, not both reactant and product. The test1.xyz file is a stand-in for your starting molecule file.
Look for the paragraph in the previous section titled “Setting up the GSM” to correctly set up the “qstart” file, which indicates the correct charge/multiplicity for the given geometry as well as the desired level of theory used for the optimization of the nodes of the string.
From here follow the instructions above outlining the use of the qsub file. All of the default settings should be fine as long as the directories are copied. For more advanced functionality and debugging please consult the advanced section below.
##Managing QSub Files for DE-GSM/SE-GSM
Add the job(s) to the qsub file:
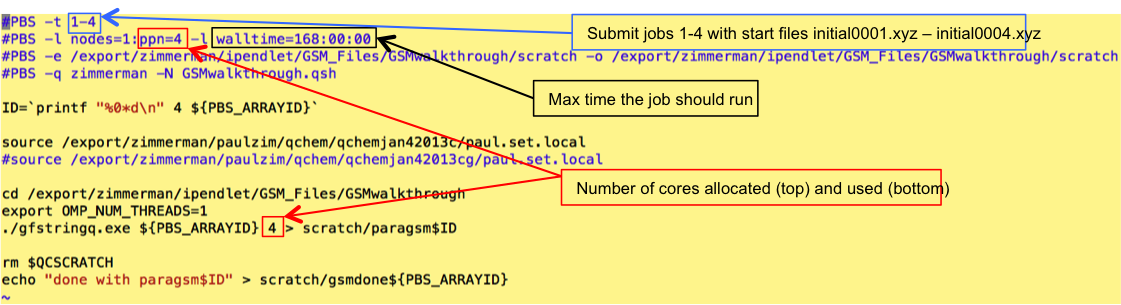
All other lines of the file (starting directories) should update automatically to the correct settings or should not be changed.
After this is complete you are able to submit your job!
Consult the “analyzing your output” section for information about GSM output.
##Analyzing Output
The key to success in analyzing the output of DE-GSM/SE-GSM jobs is using the tools provided in the copied directory (or independent tools developed for personal purposes). However, for starters let’s look at the following:
$ ./status requires the file “status” in the operating directory
$ cat stringfile.xyz00?? where stringfile.xyz00?? is the output file from the GSM/SSM
An example output from "./status" created from a random directory is as follows:

We have done our best to explain the meaning of the following outputs, but this list is incomplete. For future users, please fill in any remaining points on this output analysis that we may have missed/mistaken.
Opt_iter: 59 Displays the number of iterations that the GSM/SSM ran to complete Totalgrad: 0.073 Gradient at the transition state Gradrms: 0.007 Gradient root mean square for all nodes Tgrads: 1015 total gradients performed to complete the GSM/SSM ol(0): 0.91 overlap of the transition state with the (n+1)th Hessian vector. The number in parantheses should be either 0/1 while the overlap (0.91) should be close to (1.00)
I am not sure why there are two lines of output text from the status program. This could be residual from previous code. Potentially updating the script could be useful.
opt_iters over: totalgrad: See above gradrms: See above tgrads: See above
Important information is in the next chunk! This is where all the goodies are. The previous information is only useful for helping to debug or understand the TS run. For almost all purposes (especially when one is just starting with the software) the only needed information is below:
E: 2.7 Transition state energy in kcal/mol. In this case the TS is 2.7 kcal/mol
Erxn: -40.7 ΔH of the rxn in question. It is vital to check the product structure for SSM runs prior to reporting this energy!
Nmax: 4 Maximum energy node in the string TSnode: 4 Node optimized for TS. Usually, assuming the string grows correctly and there are no issues with multiple elementary steps in the string (happens frequently in GSM, much less likely in SSM).
-XTS- Well converged TS -TS- Moderately converged TS, could be better and might need more refinement -multistep- Multi elementary step process between starting and ending structure (GSM) -FL- The reaction proceeds uphill or downhill in energy with no transition state between reactants and products
##Advanced Options In addition to the settings listed above there are a number of control factors which can be operated by advanced users. Please do not mess with these settings unless you have experience with the method, or have been specifically told to make a change. These settings can be found in “inpfileq”. The default settings for GSM are shown below:
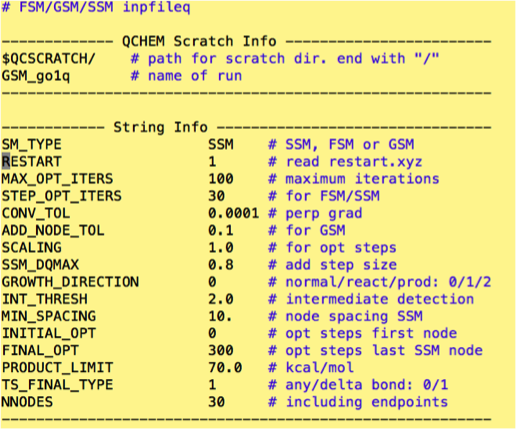
The following is a list of what these commands mean and a detailed explanation of what modifications to these settings “should” do. As this program has yet to be robustly tested on numerous structures there is the possibility for job failure if used improperly. Proceed at your own risk. Lastly, if something in the following list is found to be incorrect or incomplete, please contact the website admin to update.
SM_TYPE: Toggles between GSM, SSM and FSM (Frozen string method: don’t use it unless told to): use the indicated 3 letter combinations to utilize each of the chosen features
MAX_OPT_ITERS: Maximum iterations for each step of the GSM.
STEP_OPT_ITERS: Maximum optimizing iterations for each step of the SSM. If starting structure is not properly optimized jobs can fail if the max opt set by this variable is exceeded!
CONV_TOL: Controls the optimization threshold for nodes. Lower variables forces nodes to be optimized to a higher threshold, but can improve TS finding in difficult systems. Lower tolerances increase run time, but can improve performance if default settings fail to find the desired reaction path. (Especially useful for SSM in tricky/convoluted cases)
ADD_NODE_TOL: Tolerance variable for additional of next node in GSM. Higher numbers will afford fast growth, but decrease accuracy of reaction path identification.
SCALING: For opt steps. (To the best of my knowledge this feature controls how the step size is adjusted based on the topography of the previous optimization step. If the scaling is increased, the next step size will be the product of the DQMAX setting and the scaling variable. For more rapid adjustment based on the topography of the reaction path, increase this variable. For less automatic adjustment of the step size, decrease this variable.) For the most part, the default setting is fine.
SSM_DQMAX: Changes step size in the SSM. For the most part this setting does not need to be changed. However, when using the SSM the step size can influence the sensitivity of TS finding. Increasing this variable will decrease the sensitivity to transition states, while decreasing leads to longer times for convergence, but can be helpful if previous iterations have overshot the desired TS.
GROWTH_DIRECTION: GSM specific toggle which enables user control of growth direction. Typically the default (0) is preferred. However, this is a good toggle for debugging more difficult cases. For new users think of the location of the TS; if the TS favors the product then growth from the product with low tolerance convergence could provide better TS identification. Other combinations are also possible.
INT_THRESH: Detection threshold for intermediate during GSM and SSM growth
INITIAL_OPT: Starting structure optimization performed on the first structure of the input file.
FINAL_OPT: Max number of optimization steps performed on the last node of an SSM run.
TS_FINAL_TYPE: Determines whether rotations are considered causation for termination of the GSM or SSM run. Typically we are searching for a change in bond connectivity so this value is set to 1 for delta bond.
NNODES: Max number of nodes used for SSM and GSM. Set this number high (30) for SSM as the convergence criterion typically result in less nodes needed and too small a number for this setting will result in job failure. For GSM the typical is an odd number ranging from 9-15 with higher values being used for more robust TS finding and lower for simpler systems.
#Troubleshooting and FAQS As with most group managed software, the best resources are those that are created together. For everyone’s benefit, if you find or experience an error that is not listed below, please add it to the list! Q: How do I check the progress of specific runs of the growing string? A: Check the corresponding paragsm file.
Q: SCF fails (massively, meaning it seems that most of the runs are not making progress at a reasonable pace or they are iterating back and forth) A: Check cores - most likely need to lower the total number
Q: If all files fail and no error is returned A: Check number of cores, most likely lower. This is an uncommon case, usually the error file is somewhere. However, if all else fails this sometimes works and is a good final strategy before seeking additional help on a job.
Q: Get back error: error opening gradient file (on gsm run) A: Double check job to make sure molecule makes sense. This means that there are no missing hydrogens or obvious problems with bonding. If this is not the problem, make sure to check the qstart, inpfileq and molecule structure to make sure that charges and spin are in agreement between the files.
ZStruct/GSM interconnectivity errors: Q: Mopac deleted "significantly different" structures during a search for reasonable isomers, which resulted in only one (possibly only a few) isomers being generated. A: Mopac geometry optimizations should be turned off in constants.h: NOMOOPT=1 One would have leave mopac optimizations for manual zstruct and non-transition metal complexes.
#References
-
P. M. Zimmerman, “Growing string method with interpolation and optimization in internal coordinates: Method and examples,” Journal of Chemical Physics, 138, 184102 (2013).
-
P. M. Zimmerman, “Reliable Transition State Searches Integrated with the Growing String Method,” Journal of Chemical Theory and Computation, 9, 3043-3050 (2013).
-
P. M. Zimmerman, “Single-Ended Transition State Finding with the Growing String Method,” Journal of Computational Chemistry, 36, 601-611 (2015).