-
Notifications
You must be signed in to change notification settings - Fork 3
Commit
This commit does not belong to any branch on this repository, and may belong to a fork outside of the repository.
- Loading branch information
1 parent
7911fde
commit 65f98d3
Showing
11 changed files
with
125 additions
and
5 deletions.
There are no files selected for viewing
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
Binary file not shown.
Binary file not shown.
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
Large diffs are not rendered by default.
Oops, something went wrong.
Binary file not shown.
This file contains bidirectional Unicode text that may be interpreted or compiled differently than what appears below. To review, open the file in an editor that reveals hidden Unicode characters.
Learn more about bidirectional Unicode characters
| Original file line number | Diff line number | Diff line change |
|---|---|---|
| @@ -0,0 +1,102 @@ | ||
| # scrnabox: A pipeline for scRNA analysis under HPC | ||
| The `scrnabox.slurm` is a pipeline specifically designed for analyzing data under a High-Performance Computing (HPC) system using the Slurm Workload Manager ([slurm work load manager system](https://slurm.schedmd.com/)). It has been extensively utilized on the [Beluga](https://docs.alliancecan.ca/wiki/B%C3%A9luga), you can find comprehensive instructions on how to utilize the pipeline for [Cell Hashtags]() and [Standard scRNA-seq](), the pipeline provides detailed guidance on processing and analyzing these specific types of single-cell RNA sequencing (scRNA-seq) data. | ||
|
|
||
| Please refer to the [documentation](https://neurobioinfo.github.io/scrnabox/site/) for an explanation and of how and why to use scrnabox's pipeline. | ||
|
|
||
| ## Contents | ||
| - [Workflow analysis](#workflow-analysis) | ||
| - [Standard scRNA-seq](#standard-scRNA-seq) | ||
| - [Cell hashtags](#cell-hashtags) | ||
| - [scrnabox.slurm](#scrnaboxsvn) | ||
| - [Installing](#installing) | ||
| - [Tutorial](#tutorial) | ||
|
|
||
|
|
||
| --- | ||
|
|
||
| ## Workflow analysis | ||
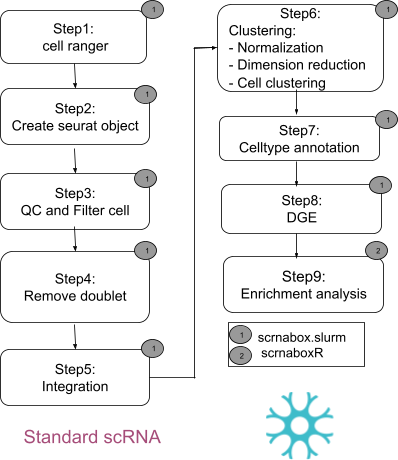
| The following figures illustrate the steps involved in analyzing scRNA-seq data using the Standard and Cell Hashtags with the scrnabox pipeline | ||
| <br /> | ||
| <br /> | ||
| <kbd> | ||
|  | ||
| </kbd> | ||
| <kbd> | ||
|  | ||
| </kbd> | ||
|
|
||
| This pipeline currently includes implementation of the standard and cell hashtags scRNA. | ||
|
|
||
| #### [Standard scRNA-seq](https://github.com/neurobioinfo/scrnabox/tree/main/README_SCRNA.md) | ||
|
|
||
| The following steps describe how to analyze scRNA-seq data using the pipeline:<br /> | ||
| - Step 1: cellranger - This step runs Cellranger on the scRNA-seq data to generate the feature-barcode matrices for each sample.<br /> | ||
| - Step 2: Seurat object - This step runs Seurat on the feature-barcode matrices obtained from step 1 to generate a Seurat object for each sample. The Seurat object contains a lot of information, and accessing [email protected] will provide a data frame with relevant information on each cell.<br /> | ||
| - Step 3: QC and filter - The Seurat object includes quality metrics that can be used to filter cells and genes against possible doublets. Metrics such as total UMI counts per cell (nCount_RNA), total number of detected features per cell (nFeature_RNA), and mitochondrial count (percent.mito) are often used.<br /> | ||
| - Step 4: Doublet removal - This step can be used to remove doublets from the data. By default, the pipeline removes doublets, but you can choose to keep them by changing the parameter from 'yes' to 'no'.<br /> | ||
| - Step 5: Integration - This step integrates multiple scRNA-seq datasets using the Comprehensive Integration of Single Cell Data (CCA) method in Seurat, Tim, et al. (2019). The pipeline identifies anchors using the FindIntegrationAnchors function and passes them to the IntegrateData function to get a single Seurat object representing all the datasets.<br /> | ||
| - Step 6: Clustering- It involves clustering the data using a k-nearest neighbor graph based on the integrated PCA. This step produces UMAP and heatmaps of unlabelled clusters. However, it is up to the user to decide on the best cluster resolution outside the pipeline by examining the output and selecting the most appropriate annotation for the clusters.<br /> | ||
| - step 7: Cluster annotate - In this step, you can annotate the clusters with known cell types or use marker genes to predict the cell type of each cluster. <br /> | ||
| - step 8: Differential gene expression (DEG)- There are multiple ways to perform differential gene expression analysis, but in this pipeline, we use the FindAllMarkers function to rank the highly differentially expressed genes in each cluster, which allows us to identify genes that are significantly differentially expressed between each cluster and the rest of the cells. From there, we can define contrasts to run statistical tests and investigate the phenotype and genotypes of each cluster.<br /> <br /> | ||
| The Step 1 - Step 8 can be done using [scrnabox.slurm](https://github.com/neurobioinfo/scrnabox/tree/main/README_SCRNA.md) in the HPC system ([slurm work load manager system](https://slurm.schedmd.com/)). | ||
| - step 9, Enrichment analysis: in this step, we obtain a list of significant genes using enrichment methods. The step 9 can be done using scrnaboxR, see [Practice](https://github.com/neurobioinfo/scrnabox/blob/main/tutorial/practice.md). | ||
|
|
||
|
|
||
| #### [Cell Hashtags](https://github.com/neurobioinfo/scrnabox/tree/main/README_HTO.md) | ||
|
|
||
| The following steps explain how to analyze the Hashtag oligonucleotide (henceforth referred to as HTO) | ||
| - Step 1: cellranger - This step runs Cellranger on the scRNA-seq data to generate the feature-barcode matrices for each sample. | ||
| - Step 2: Seurat object - This step runs Seurat on the feature-barcode matrices obtained from step 1 to generate a Seurat object for each sample. The Seurat object contains a lot of information, and accessing [email protected] will provide a data frame with relevant information on each cell. | ||
| - Step 3: QC and filter - The Seurat object includes quality metrics that can be used to filter cells and genes against possible doublets. Metrics such as total UMI counts per cell (nCount_RNA), total number of detected features per cell (nFeature_RNA), and mitochondrial count (percent.mito) are often used. | ||
| - Step 4: Demuplixing- In this step, demultiplexing is performed to separate the reads in the sequencing run according to their sample of origin, based on their barcode information. The pipeline includes an option to remove doublets and negative cells during a later step after quality control and filtering. | ||
| - Step 5: Integration - This step integrates multiple scRNA-seq datasets using the Comprehensive Integration of Single Cell Data (CCA) method in Seurat, Tim, et al. (2019). The pipeline identifies anchors using the FindIntegrationAnchors function and passes them to the IntegrateData function to get a single Seurat object representing all the datasets. | ||
| - Step 6: Clustering- It involves clustering the data using a k-nearest neighbor graph based on the integrated PCA. This step produces UMAP and heatmaps of unlabelled clusters. However, it is up to the user to decide on the best cluster resolution outside the pipeline by examining the output and selecting the most appropriate annotation for the clusters. | ||
| - step 7: Cluster annotate - In this step, you can annotate the clusters with known cell types or use marker genes to predict the cell type of each cluster. | ||
| - step 8: Differential gene expression (DEG)- There are multiple ways to perform differential gene expression analysis, but in this pipeline, we use the FindAllMarkers function to rank the highly differentially expressed genes in each cluster, which allows us to identify genes that are significantly differentially expressed between each cluster and the rest of the cells. From there, we can define contrasts to run statistical tests and investigate the phenotype and genotypes of each cluster. | ||
|
|
||
| The Step 1 - Step 8 can be done using [scrnabox.slurm](https://github.com/neurobioinfo/scrnabox/tree/main/README_HTO.md) in the HPC system ([slurm work load manager system](https://slurm.schedmd.com/)). | ||
| - step 9, Enrichment analysis: in this step, we obtain a list of significant genes using enrichment methods. The step 9 can be done using scrnaboxR, see [Practice](https://github.com/neurobioinfo/scrnabox/blob/main/tutorial/practice.md). | ||
|
|
||
|
|
||
|
|
||
| ## Installing | ||
| The package is written in the bash, so it can be used with any slurm system. To download | ||
| `scrnabox.slurm` run the below comments | ||
| ``` | ||
| wget https://github.com/neurobioinfo/scrnabox/releases/download/v0.1.0/scrnabox.slurm.zip | ||
| unzip scrnabox.slurm.zip | ||
| ``` | ||
|
|
||
| To obtain a brief guidance of the pipeline, execute the following code. | ||
| ``` | ||
| bash ./scrnabox.slurm/launch_scrnabox.sh -h | ||
| ``` | ||
|
|
||
| `scrnabox.slurm` needs `R` and `cellranger`. For the R, you need to install | ||
| `'Seurat','ggplot2', 'dplyr', 'foreach', 'doParallel', 'Matrix', 'DoubletFinder','cowplot','clustree'`. Then install `'scrnaboxR'`: | ||
| ``` | ||
| devtools::install_github("neurobioinfo/scrnabox/scrnaboxR") | ||
| ``` | ||
| The `'scrnaboxR'` is an R package that provides a collection of functions for conducting enrichment analysis and other analyses associated with single-cell RNA sequencing (scRNA-seq) data. It serves as a companion to scrnabox, offering a range of tools and functionalities to enhance scRNA-seq data analysis. You need to add the R info in `scrnabox_config.ini`, you can define the path of R library in `R_LIB_PATH=`, version of R in `R_VERSION`, you can add the path of `cell ranger`in `MODULECELLRANGER` | ||
|
|
||
|
|
||
| ## Tutorial | ||
| You can find the details of how to use the pipeline in the | ||
| the [documentation](https://neurobioinfo.github.io/scrnabox/site/). | ||
|
|
||
|
|
||
| --- | ||
| #### Contributing | ||
| This is an early version, any contribute or suggestion is appreciated, you can directly contact with developers: [Saeid Amiri](https://github.com/saeidamiri1), [Michael Fiorini](https://github.com/fiorini9), or [Rhalena Thomas](https://github.com/RhalenaThomas). | ||
|
|
||
| #### Changelog | ||
| Every release is documented on the [GitHub Releases page](https://github.com/neurobioinfo/scrnabox/releases). | ||
|
|
||
| #### License | ||
| This project is licensed under the MIT License - see the [LICENSE.md](https://github.com/neurobioinfo/scrnabox/blob/main/LICENSE) file for details. | ||
|
|
||
| #### Acknowledgement | ||
| The pipeline is done as part Dark Genome project, it is written by [Saeid Amiri](https://github.com/saeidamiri1) with associate of Rhalena Thomas, Sali Farhan, and Michael Fiorini at Neuro Bioinformatics Core. Copyright belong MNI BIOINFO CORE (https://github.com/neurobioinfo). | ||
|
|
||
| **[⬆ back to top](#contents)** | ||
|
|